mukopolysacharidóza je spoločný termín pre choroby lyzozomálneho skladovania, ktoré sú založené na skladovaní glykozaminoglykánov. Pri všetkých chorobách sa vyvíjajú podobné príznaky a formy. Závažnosť syndrómov sa veľmi líši.
Čo je mukopolysacharidóza?

© Sebastian Kaulitzki - stock.adobe.com
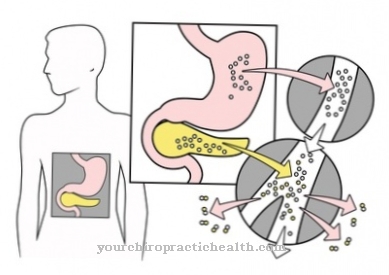
mukopolysacharidóza neexistuje jediná choroba. Termín mukopolysacharidóza je súhrnný pojem pre veľké množstvo chorôb z ukladania, ktoré sú založené na poruchách ukladania glykozaminoglykánov (GAG) v lyzozómoch buniek. Ukladanie prebieha postupne, pretože ukončenie spojení nefunguje.
Všetky mukopolysacharidózy sú genetické. Každému ochoreniu chýba špecifický enzým, ktorý katalyzuje rozklad príslušného GAG. Všetky mukopolysacharidózy sú veľmi zriedkavé choroby a často vykazujú podobné priebehy. Ak sa nelieči, stále sa zvyšujúce depozity ničia bunky. Pri tomto procese sa orgány ničia. Ochorenie môže začať v detstve aj v dospelosti.
Mukopolysacharidóza môže byť spôsobená štyrmi rôznymi skupinami glykozaminoglykánov:
- Heparan sulfát
- Keratan sulfát
- Chondroitín sulfát
- Dermatan sulfát.
Všetky glykozaminoglykány pozostávajú z polysacharidového reťazca, ktorý je spojený s proteínom. Sacharidová zložka tvorí 95 percent a proteínová zložka päť percent molekulovej hmotnosti. V závislosti od toho, ktorý glykozaminoglykán a ktorý enzým je ovplyvnený, sa mukopolysacharidózy môžu rozdeliť do šiestich rôznych hlavných foriem: Patria sem Hurlerova / Scheieho choroba (MPS I), Hunterova choroba (MPS II), Sanfilippoova choroba (MPS III), Morquiova choroba ( MPS IV), Maroteaux-Lamyho choroba (MPS VI) a Slyho choroba (MPS VII). Všetky typy majú ťažké a mierne formy.
príčiny
Príčinou všetkých mukopolysacharidóz je zvýšené ukladanie glykozaminoglykánov (GAG) v lyzozómoch buniek. Degradácia zodpovedajúcich biopolymérov je narušená. Pre každú jednotlivú poruchu chýba určitý enzým alebo tento enzým funguje nesprávne. Pre každý enzým môže existovať niekoľko mutácií. Dedičnosť zodpovedajúcej mutácie môže byť autozomálne recesívna, autozomálne dominantná alebo x-spojená recesívna.
Pretože enzymatický proces obvykle zahrnuje niekoľko reakčných krokov, môže byť teoreticky mutovaných niekoľko enzýmov pre rovnaký glykozaminoglykán. Príznaky poruchy by boli rovnaké alebo podobné.
- na MPS I, Hurlerova alebo Scheieho choroba, enzým alfa-l-iduronidáza je chybný.
- MPS II predstavuje Huntersov syndróm s defektnou iduronát-2-sulfatázou.
- Syndróm Sanfilippo (MPS III) možno rozdeliť do niekoľkých podtypov. V tomto stave môže byť ovplyvnených niekoľko enzýmov.
- Morquiova choroba (MPS IV) je spôsobená defektnou P-galaktozidázou.
- Pri syndróme Maroteaux-Lamy (MPS VI) je to N-acetyl-galaktozamín-4-sulfát sulfatáza.
- Slyho choroba (MPS VII) je spôsobená defektnou ß-glukuronidázou. Ak sú zodpovedajúce glykozaminoglykány uložené v lyzozómoch, tieto sa zväčšujú a zväčšujú.
Bunky sa tiež zväčšujú, pretože vyžadujú viac a viac miesta pre nedegradované GAG. Je to zrejmé aj pri rozširovaní mnohých orgánov. Typickým príznakom je neustále zväčšovanie pečene a sleziny. Ak sa ochorenie pri skladovaní nelieči, vedie k postupnému ničeniu orgánov k úmrtiu.
Príznaky, choroby a príznaky
Príznaky sú podobné pre všetky choroby. Existujú ťažké a mierne formy. Mierny priebeh však znamená, že choroba postupuje pomalšie. Záverečný kurz je vždy rovnaký. Dochádza k progresívnej deformácii kostrového systému, kĺbových kontraktúr, hrubovaniu rysov tváre a zväčšeniu pečene a sleziny.
Mentálne a motorické zručnosti sa v krátkodobom alebo dlhodobom horizonte znižujú. Pri závažných formách porúch sú klinické obrazy veľmi podobné. V počiatočnom štádiu sa vyskytujú pupočné a trieslové prietrže, srdcové problémy a infekcie dýchacích ciest. V priebehu času sa zúženie dýchacích ciest a zväčšenie mandlí a mandlí vyvinuli v masívne problémy so spánkovým apnoe.
Diagnóza a priebeh choroby
Mukopolysacharidózy sa môžu diagnostikovať vyšetrením vylučovaných glykozaminoglykánov v moči. Pri mukopolysacharidóze sa hodnoty vždy zvyšujú. Môže byť tiež stanovená aktivita podozrivého defektného enzýmu v leukocytoch alebo fibroblastoch. Určitý spôsob vylučovania glykozaminoglykánov vedie k podozreniu na zodpovedajúci enzým, ktorý sa potom skúma.
komplikácie
Z dôvodu mukopolysacharidózy postihnuté osoby trpia rôznymi malformáciami a kostnými ťažkosťami. Vyskytujú sa deformácie, ktoré môžu významne obmedziť každodenný život pacienta. Kíby sú spravidla ovplyvnené aj mukopolysacharidózou, takže pohyb pacienta je obmedzený.
Najmä deti sú postihnuté a trpia výrazne oneskoreným vývojom, takže v dospelosti sa môžu vyskytnúť rôzne následné škody. Nie je neobvyklé, že mukopolysacharidóza spôsobuje problémy so srdcom alebo dýchanie. V najhoršom prípade môže náhla srdcová smrť viesť k smrti dotknutej osoby. V dôsledku ťažkostí s dýchaním trpia pacienti únavou a únavou.
Odolnosť postihnutých tiež výrazne klesá. Nie je neobvyklé, že dýchacie ťažkosti v noci vedú k problémom so spánkom a tým k depresii. Mukopolysacharidóza výrazne znižuje kvalitu života pacienta. Príčinná liečba tohto ochorenia bohužiaľ nie je možná. Postihnuté osoby sú preto pri liečbe príznakov závislé od darcov kostnej drene. Neexistujú žiadne osobitné komplikácie. Vo väčšine prípadov sú však pacienti závislí od celoživotnej terapie.
Kedy by ste mali ísť k lekárovi?
Zmeny a odchýlky v štruktúre tela naznačujú poškodenie zdravia. Návšteva lekára je nevyhnutná, len čo sa vyskytnú trvalé optické zvláštnosti alebo ak dotknutá osoba má problémy s úmyslom optimalizovať svoju polohu.Opuch kĺbov, zmeny v tvárových rysoch alebo zväčšenie hrudníka by mal lekár intenzívne vyšetriť, aby bolo možné stanoviť diagnózu. Ak existujú obmedzenia v pohybe, nepravidelnosti v každodennej dobrovoľnej kontrole a pokles fyzickej a duševnej výkonnosti, vyžaduje sa lekár. V prípade porúch srdcového rytmu, problémov s dýchaním alebo prerušením nočného spánku by sa malo konzultovať s lekárom.
Opuch v hrdle, pocit zovretia v hrdle, poruchy pri prehĺtaní a zmeny vo vokalizácii sa považujú za znepokojujúce. Musí ich vyšetriť lekár, aby sa zmiernili príznaky. Ak dotknutá osoba trpí väčším počtom infekcií, ak sa schopnosť sústrediť sa a venovať pozornosť alebo opakovane objaví pupočná alebo trieslová prietrž, lekár by mal byť o týchto zisteniach informovaný.
Mali by sa vyšetriť a ošetriť náhle škvrny na koži, zožltnutie kože a vnútorný nepokoj. Hneď ako je v tele bolesť, znížená kvalita života a problémy so správaním, je potrebný lekár. Ak existuje riziko dýchavičnosti, je potrebná záchranná služba. Aby sa zabránilo tomuto akútnemu stavu, je potrebné sa poradiť s lekárom čo najskôr.
Terapia a liečba
Kauzálna terapia dnes ešte nie je možná. Vo výskumných projektoch však existuje niekoľko prístupov k budúcej génovej terapii týchto chorôb. V tejto oblasti bohužiaľ momentálne nie sú hmatateľné výsledky. V Barcelone sa však má začať klinická štúdia génovej terapie Hurlerovej choroby. Pri niektorých formách mukopolysacharidóz sa prenosy kostnej drene ukázali ako účinné v jednotlivých prípadoch. Týka sa to napríklad Hunterovej choroby, Hurlerovej choroby alebo Sanfilippoho choroby.
Týmto prenosom kostnej drene sa choré kmeňové bunky vymieňajú za zdravé kmeňové bunky od darcu. Toto umožňuje organizmu dostatočne obnoviť chýbajúci enzým. V mnohých prípadoch sa oplatí aj náhradná enzýmová terapia. Táto substitučná terapia sa však musí vykonávať po celý život. Existujú však aj prípady, keď sľubné terapie už nie sú možné. Ide tu však o vykonanie symptomatickej liečby.
Svoje lieky nájdete tu
➔ Lieky proti bolestiVýhľad a predpoveď
Ďalší vývoj u pacientov s mukopolysacharidózou sa musí hodnotiť individuálne. Tento výraz je kolektívny pojem pre rôzne choroby z ukladania. Tieto sú prítomné v rôznom stupni u každého pacienta a sú individuálne vyjadrené svojou intenzitou. Ak sa lekárska starostlivosť nezačne, vnútorné orgány všetkých postihnutých sa v priebehu života postupne ničia. To má za následok skrátenie priemernej očakávanej životnosti.
Pri včasnej diagnóze je možné vypracovať osobne optimalizovanú terapiu. Súvisí to so zdravotnými požiadavkami a existujúcimi sťažnosťami pacienta. Na dosiahnutie stabilného zlepšenia zdravia je nevyhnutná dlhodobá liečba. Môžu sa vyskytnúť chirurgické zákroky, z ktorých každý je spojený s rôznymi rizikami a vedľajšími účinkami. Ak operácia pokračuje bez akýchkoľvek ďalších komplikácií, príznaky sa zvyčajne zmiernia.
V priebehu života sa však môžu vyskytnúť nežiaduce udalosti a neúspechy. V individuálnych prípadoch iba transplantácia kostnej drene môže zlepšiť celkovú kvalitu života. Vzhľadom na celkové okolnosti pacient trpí veľkým emocionálnym a psychickým stresom. Normálny každodenný život nie je kvôli príznakom často možný. Môžu sa vyskytnúť psychologické komplikácie a viesť k ďalšiemu zhoršeniu situácie.
prevencia
Pretože mukopolysacharidózy sú dedičné choroby, prevencia nie je možná. V prípade existujúceho ochorenia môže byť úspech liečby zaistený včasnou liečbou. Okrem toho je potrebné neustále monitorovanie funkcie pľúc a srdca. Ak sa už v rodine vyskytli prípady mukopolysacharidózy, riziko ochorenia sa dá vyhodnotiť pomocou genetického poradenstva, ak si rodina želá mať deti.
domáce ošetrovanie
Vo väčšine prípadov mukopolysacharidózy má pacient len niekoľko možností následnej starostlivosti, takže postihnutá osoba by sa mala v prvom rade poradiť s lekárom v ranom štádiu. Ďalším komplikáciám je možné predchádzať iba včasným odhalením a liečbou tohto ochorenia, takže akonáhle sa objavia prvé príznaky a príznaky, kontaktujte lekára.
Vo väčšine prípadov sú postihnutí závislí od chirurgických zákrokov, ktoré môžu zmierniť a obmedziť príznaky. Avšak, pretože mukopolysacharidóza je genetické ochorenie, zvyčajne sa nedá úplne vyliečiť.
Z tohto dôvodu by sa dotknutá osoba mala najprv poradiť s lekárom, ak chce mať deti, aby sa zabránilo opakovaniu tohto ochorenia u detí. Počas liečby je často veľmi dôležité mať podporu rodiny. To tiež môže zabrániť depresii a iným psychickým rozrušeniam. Mukopolysacharidóza môže viesť k zníženiu očakávanej dĺžky života postihnutej osoby, pričom ďalší priebeh závisí vo veľkej miere od času diagnózy.
Môžete to urobiť sami
Možnosti svojpomoci s mukopolysacharidózou sú obmedzené na zmiernenie príznakov, a tým na zlepšenie kvality života. Svojpomocné skupiny sa ukázali ako veľmi užitočné, pretože výmena s ostatnými rodičmi odhaľuje cenné tipy a často môže zmierniť obavy a obavy a poskytnúť pozitívnejší pohľad na budúcnosť.
Sprievod k fyzioterapii, ergoterapii, rečovej terapii a iným formám liečby, ktoré sa v domácom prostredí často prehlbujú, je dnes neoddeliteľnou súčasťou života.
S cieľom čo najjednoduchšieho života pre seba a postihnuté dieťa je vhodné čo najskôr sprístupniť životné prostredie osobám so zdravotným postihnutím. S rastúcim vekom a hmotnosťou dieťaťa sa výškovo nastaviteľné detské postieľky pre opatrovateľa stávajú veľkou fyzickou úľavou. Výstražné zariadenia na epilepsiu a ďalšie technické pomôcky umožňujú najlepšiu možnú bezpečnosť aj v noci a uvoľňujú rodičov v noci, aby mohli pohodlnejšie spať.
Vedenie denníka symptómov môže lekárovi pomôcť rozpoznať nové príznaky a prípadne opraviť liečbu existujúcich príznakov, pretože lieková terapia často nevykazuje požadovaný, ale opačný účinok.
Keďže choroba je pre príbuzných veľmi náročná, musia si sami vytvoriť malý priestor na dobitie svojich batérií. To môže zahŕňať liečebné postupy, preventívnu starostlivosť alebo neskôr dovolenku v hospici.

.jpg)






.jpg)

.jpg)